molecularDNA macro files
Macro files define simulations in Geant4. The molecularDNA example contains a number of additional commands that allow DNA damage simulations to take place. The full list of commands is written in configuration.
Table of contents
- A basic macro file
- Set up the world
- Set up the DNA geometry
- Define the chromosomes
- Set up the damage model
- Set up the beam
- Set up analysis rules
- Run the simulation
A basic macro file
Let’s start with a basic macro file:
# First off, we set the size of the world
/world/worldSize 3 um
# Next we define the geometry
/dnageom/placementSize 50 50 50 nm
/dnageom/placementVolume turn geometries/1strand_50nm_turn.txt
/dnageom/placementVolume turntwist geometries/1strand_50nm_turn.txt true
/dnageom/placementVolume straight geometries/1strand_50nm_straight.txt
/dnageom/fractalScaling 50 50 50 nm
/dnageom/definitionFile geometries/hilbert1.txt
# We define our "chromosomes"
/chromosome/add cell sphere 200 0 0 0 nm
# Geometry related damage parameters
/dnageom/radicalKillDistance 4 nm
/dnageom/interactionDirectRange 6 angstrom
# We define the damage model
/dnadamage/directDamageLower 17.5 eV
/dnadamage/directDamageUpper 17.5 eV
/dnadamage/indirectOHBaseChance 1.0
/dnadamage/indirectOHStrandChance 0.4
/dnadamage/inductionOHChance 0.00
/dnadamage/indirectHBaseChance 1.0
/dnadamage/indirectHStrandChance 0.4
/dnadamage/inductionHChance 0.00
/dnadamage/indirectEaqBaseChance 1.0
/dnadamage/indirectEaqStrandChance 0.4
/dnadamage/inductionEaqChance 0.00
# And then we initialize our run
/run/initialize
# The General Particle Source is used to define a beam
/gps/particle proton
/gps/ang/type iso
/gps/energy 100 keV
/gps/pos/type Point
/gps/pos/centre 51 51 51 nm
# Set any analysis parameters
/analysisDNA/fileName my_file.root
# And the simulation can run!
/run/beamOn 1
That’s a lot to digest. But we can break down the macro file into a few components:
- Set up the world
- Define the geometry
- Define the regions of interest (chromosomes)
- Define the damage model
- Set up the beam
- Set any analysis rules
- Run the simulation
Set up the world
The first command to run is to set the size of the world. The simulation runs in a square world and you can set the side length as below:
/world/worldSize 3 um
The world is entirely made of liquid water unless you also specify a cell (by its semi-major axes).
/cell/radiusSize 1 1 0.5 um
Cells modify the default behaviour by placing a water containing cell into a vacuum-filled world.

Set up the DNA geometry
There are two important notions when we define the geometry, placement definition and the geometry definition, often called the fractal definition as it is often seeded with a fractal.
- Placement definition defines the small scale structure of the simulation.
- Geometry definition defines the large scale structure of the simulation.
This is illustrated below, where turned and straight segments are used to build a horse-shoe shape:
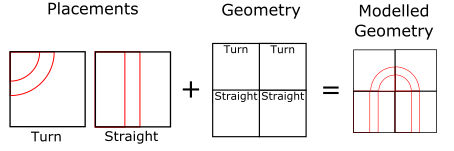
So how does all this work? Let’s break down the definitions above:
The first things we define are the sizes of our placement volumes. Here we use 50 nm side length cubes, but these can be rectangular prisms of any shape.
/dnageom/placementSize 50 50 50 nm
In this example there are three placement definitions for a straight section, a turned section, and a turned with a 90˚ twist section.
/dnageom/placementVolume turn geometries/1strand_50nm_turn.txt
/dnageom/placementVolume turntwist geometries/1strand_50nm_turn.txt true
/dnageom/placementVolume straight geometries/1strand_50nm_straight.txt
These are referenced by the their names turn, straight and turntwist in the geometry definition file which is loaded as follows:
/dnageom/definitionFile geometries/hilbert1.txt
This particular file handles the space in integer units, so that we can scale them using the command, which allows the definition file to be scaled to the size of the placement volumes we use in this example.
/dnageom/fractalScaling 50 50 50 nm
Define the chromosomes
In the molecularDNA example, you can think of chromosomes as regions of interest for analysis. Currently, cylindrical, spherical and elliptical chromosome shapes can be defined, which are mapped onto the placement volumes defined by the large scale geometry.
The below image shows, for example how two chromosomes can be defined to yield two regions of interest, overlaid on the grid which defines the overall geometry. Where chromosome regions overlap, energy depositions will be recorded in both.

Importantly, the DNA geometry is only placed inside chromosomes. Using this, a square geometry can be coerced to a circular form, by placing a spherical chromosome as we do here:
/chromosome/add cell sphere 200 0 0 0 nm
The chromosome name is cell, its shape is sphere. The radius 200. The position : 0 0 0. The unit: nm nanometer.
The arguments for a cylinder are:
- name cyl radius height x y z unit
- name cyl radius height x y z unit rx ry rz (rotations)
The arguments for a ellipse are:
- name ellipse sx sy sz x y z unit
- name ellipse sx sy sz x y z unit rx ry rz
Note that dimensions (sx, sy, sz) are semi-major axes and rotations are in degrees.
Set up the damage model
A number of parameters are used to determine how DNA damage works in the simulation. The first of these determine how the geometry in particular influences the simulation.
/dnageom/radicalKillDistance 4 nm
/dnageom/interactionDirectRange 6 angstrom
The radical kill distance tells the simulation to kill all chemistry tracks further than 4 nm from the DNA. This parameter is an implicit bound on scavenging. It basically assumes all chemical radicals that need to diffuse 4 nm to react with the DNA will be scavenged before they are able to interact chemically with the DNA molecule.
The direct interaction range describes to what radius direct (physics-driven) energy depositions should be ascribed to a DNA molecule. Here, only energy depositions with 6 Å can contribute a direct strand break.
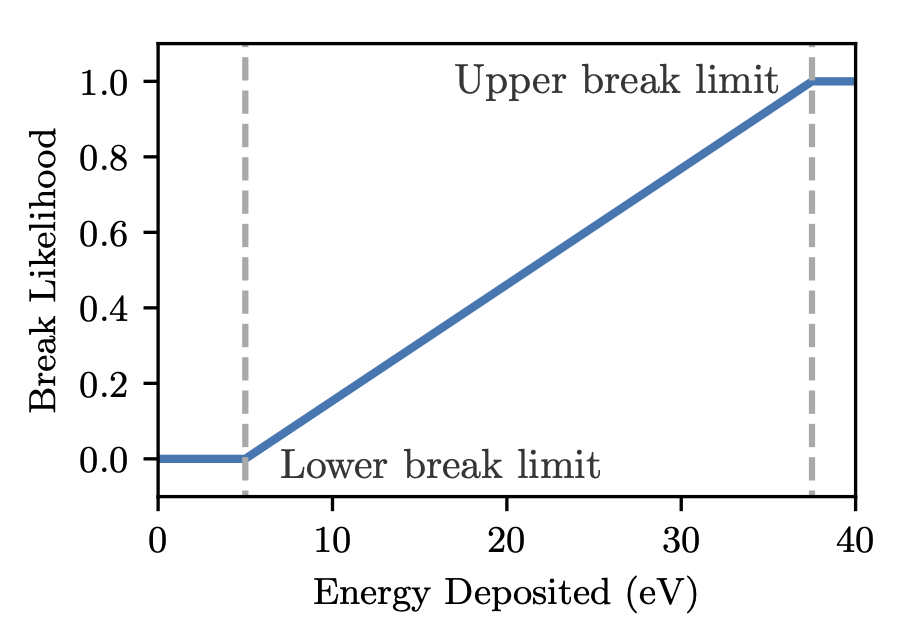
The next part of the damage model handles how direct strand breaks are calculated. The below snippet defines a step function where a cumulative deposition of 17.5 eV or more in one event will cause a break.
/dnadamage/directDamageLower 17.5 eV
/dnadamage/directDamageUpper 17.5 eV
The lower and upper values here describe a broken linear function where:
- Energy deposition below directDamageLower never causes a break
- Energy deposition above directDamageUpper always causes a break
- Between these bounds, the likelihood of a break rises uniformly
An example of this, for a lower bound of 5 eV and an upper bound of 37.5 eV is shown below
Next, we define the likelihood of chemical damage occurring for different reactions as below:
/dnadamage/indirectOHBaseChance 1.0
/dnadamage/indirectOHStrandChance 0.4
/dnadamage/inductionOHChance 0.0
Indirect damage here is what is typically discussed in most papers, it is the likelihood of a chemical reaction occurring between either •OH (in this case) and either a base or strand molecule. A lot of research only considers reactions between radicals and strands as leading to strand breaks, and this simulation considers that all reactions between a strand and a radical cause a break.
On the other hand, it’s rarely considered that an interaction between a radical and a base can lead to a strand break. If, for whatever reason, you want to model this, you can use the induction chance. This is the probability that base damage leads to a strand break.
For most simulations, all that is important are the radical + strand break chances, which are set by:
/dnadamage/indirectOHStrandChance 0.4
/dnadamage/indirectHStrandChance 0.4
/dnadamage/indirectEaqStrandChance 0.4
for the •OH, H+ and eaq radicals respectively.
Set up the beam
The simulation allows the Geant4 General Particle Source be used to define the beam. You can read the documentation here.
The source here defines a 100 keV proton point source emitting isotropically.
# The General Particle Source is used to define a beam
/gps/particle proton
/gps/ang/type iso
/gps/energy 100 keV
/gps/pos/type Point
/gps/pos/centre 51 51 51 nm
Set up analysis rules
The commands under analysisDNA allow the analysis to be modified. Notably, you’ll want to set the output file name:
/analysisDNA/fileName my_file.root
Run the simulation
Particles can be fired using the /run/beamOn command. If you haven’t already run /run/initialize, the initialisation process will also occur here. Be advised that this can take many hours to build a complex DNA geometry due to how the simulation has been designed and the current constraints of Geant4 as it optimises navigation. It is advised that a test on a small geometry is run before moving to a human cell-sized geometry.