Geometry model
Table of contents
Design principle
The molecularDNA application was made to make it easy to define a DNA geometry, and then place it repeatedly to model a complex geometry at large scales, as below.
There are two important notions when we define the geometry, placement definition and the geometry definition, often called the fractal definition as it is often seeded with a fractal.
- Placement definition defines the small scale structure of the simulation.
- Geometry definition defines the large scale structure of the simulation.
Some examples of how to make these files are available on Github, and examples can be found from the Available geometries
DNA placements
DNA placements are the low level building block of the DNA geometry and show how the DNA sits in a given prism.
A simple example of this is below, containing 14 base pairs (grey), alongside the sugar (red) and phosphate (yellow) molecules in the chain.
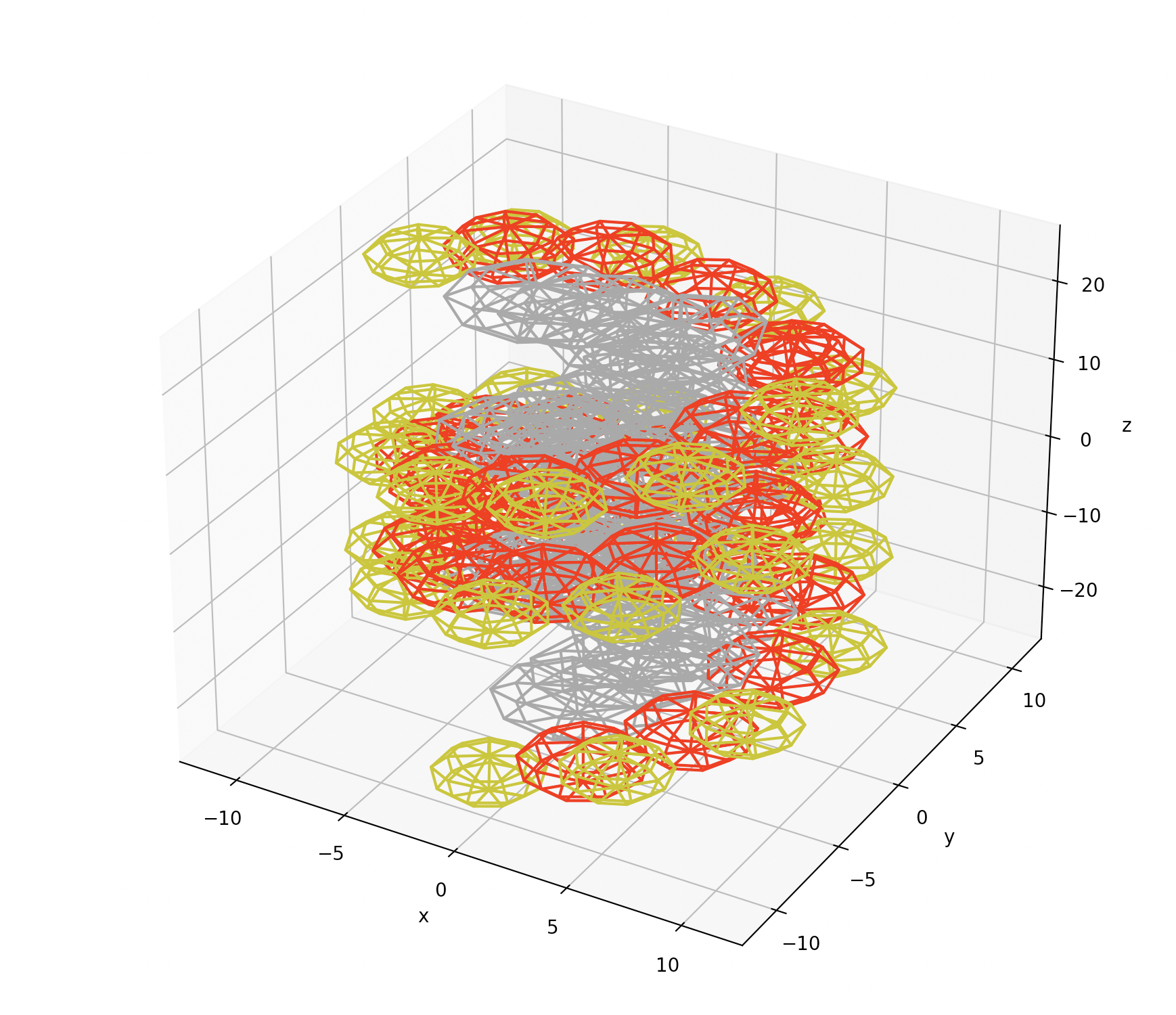
These files are specified as either 11 or 14 column ASCII text files with the schema below:
- MOLECULE_NAME - name of the molecule
- SHAPE optional - shape of the molecule (sphere or ellipse)
- CHAIN_ID - index for the chain
- STRAND_ID - index for the strand (either left or right side of the DNA molecule)
- BP_INDEX - index of the base pair in the volume (per chain)
- SIZE_X optional - semi-major x axis of ellipse (Å)
- SIZE_Y optional - semi-major y axis of ellipse (Å)
- SIZE_Z optional - semi-major z axis of ellipse (Å)
- POS_X - x position in volume (Å)
- POS_Y - y position in volume (Å)
- POS_Z - z position in volume (Å)
- ROT_X - rotation around the X-axis
- ROT_Y - rotation around the Y-axis
- ROT_Z - Rotation around the Z-axis
Distances are specified in Angstrom, while angles are specified in radians (and correspond to the three Euler angles).
Specifying molecule sizes
The molecule size columns are optional, as they can either fall back onto the default sizes or be set to custom sizes in the macro definition via:
/dnageom/useCustomMoleculeSizes
/dnageom/moleculeSize name x y z units
Note that molecule name matches are case insensitive.
The default molecule names and sizes are:
| Molecule name | X semi-major axis (Å) | Y semi-major axis (Å) | Z semi-major axis (Å) |
|---|---|---|---|
| phosphate | 2.282354 | 2.282354 | 2.282354 |
| sugar | 2.632140 | 2.632140 | 2.632140 |
| guanine | 3.631503 | 3.799953 | 1.887288 |
| cytosine | 3.597341 | 3.066331 | 1.779361 |
| adenine | 3.430711 | 3.743504 | 1.931958 |
| thymine | 4.205943 | 3.040448 | 2.003359 |
| histone | 25 | 25 | 25 |
Adding multiple chains
A simple way to increase the density of DNA in simulations is to place multiple chains of DNA in each placement volume. Multiple ‘chains’ can be placed in the one placement volume as shown below, provided that they each have a different chain_id in the data file. Here, we show graphically what a 1-chain and 4-chain placement volume could look like.
The simulation platform supports 1, 4 and 8 chains. It will join chains together correctly even when placement volumes are rotated along the lines of the image shown below.
Defining DNA placements
Placement files should be defined with either of the following two (space seperated) schemas:
Specifying size for every molecule
NAME SHAPE CHAIN_ID STRAND_ID BP_INDEX SIZE_X SIZE_Y SIZE_Z POS_X POS_Y POS_Z ROT_X ROT_Y ROT_Z
Not specifying size for every molecule
NAME CHAIN_ID STRAND_ID BP_INDEX POS_X POS_Y POS_Z ROT_X ROT_Y ROT_Z
Spatial units are Angstroms, angles are in radians. Molecule names cannot contain spaces.
The program assumes that the file is ordered first by base pair, then strand, then chain as below, keeping the order phosphate, sugar then base. Histones when specified can be placed anywhere in the file however (they are often at the start or end).
NAME ... CHAIN_ID STRAND_ID BP_INDEX
Phosphate 0 0 0
Sugar 0 0 0
Base 0 0 0
Phosphate 0 1 0
Sugar 0 1 0
Base 0 1 0
Phosphate 0 0 1
Sugar 0 0 1
Base 0 0 1
Phosphate 0 1 1
Sugar 0 1 1
Base 0 1 1
Phosphate 0 0 2
Sugar 0 0 2
Base 0 0 2
Phosphate 0 1 2
Sugar 0 1 2
Base 0 1 2
Phosphate 1 0 1
Sugar 1 0 1
Base 1 0 1
The order phosphate - sugar - base must always be kept. Each chain is specified completely before a new chain starts, and base pair sections are specified in their entirety before a new base pair section is specified.
This is done to enable the file to be read in such that their is a fixed distance between any molecule and its neighbours. This facilitates the cutting of certain shapes within Geant4 so that each shape can be represented with the maximum volume possible.
The program will assume the neighbouring molecules for a given sugar, phosphate or base are:
- Base Pair, strand ID 0: sugar at :-1, base at :+3, base at :-6
- Base Pair, strand ID 1: sugar at :-1, base at :-3, base at :-6
- Sugar: phosphate at L-1
- Phosphate: sugar at L+5
Once defined, a placement volume can be loaded using the following command:
/dnageom/placementVolume name path twist
For example:
/dnageom/placementVolume turntwist geometries/turned_twisted_solenoid_750.txt true
Considerations for base pairs
When building the geometry, you’ll probably consider a base pair in an unrotated space and then rotate the base pair to produce the chain you desire, as well as the standard double helix.
Our geometries have been built and tested with the primary axis of the double helix in a straight DNA segment being the Z-axis. Similarly, a base pair on its own is built so that its height (the 3.4 Å-long axis) is the Z-axis. We rely on this assumption to know that, in a frame of reference prior to any rotation, a base pair should be roughly 3.4 Å in height.
This assumption is used to help stop molecule placements intersecting each other. If you are developing geometries alongside the Python package FractalDNA, these changes should be done automatically.
Geometry placements
The Geometry definition files describe how the DNA placements should be put into the simulation world.
Often it will define tesselating cubes that join DNA together in a snake-like pattern, though it can also be used to place non-connected DNA elements (e.g. this parameter study).
Three control parameters are useful when placing the geometry:
/analysisDNA/fragmentGap 0will treat all placements as separate, preventing any joining between strands./dnageom/setVoxelPlacementAnglesAsMultiplesOfPiwill load the Euler angles for placement volumes as multiples of pi./dnageom/fractalScaling X Y Z nmwill scale the distances in the geometry file by the values specified.- e.g.
/dnageom/fractalScaling 1 1 1 nmwill mean that the geometry file is in units of nm.
- e.g.
The schema for the geometry placement files is as follows, with rotations being the Euler angles.
- IDX - placement index
- KIND - placement name (should correspond to a DNA volume definition)
- POS_X - X position (mm if not scaled)
- POS_Y - Y position (mm if not scaled)
- POS_Z - Z position (mm if not scaled)
- EUL_PSI - placement rotation (psi)
- EUL_THETA - placement rotation (theta)
- EUL_PHI - placement rotation (phi)
An example file might look like this.
#IDX KIND POS_X POS_Y POS_Z EUL_PSI EUL_THETA EUL_PHI
0 straight 0.0 0.0 0.0 0.0 -0.0 0.0
1 turn 0.0 0.0 1.0 0.0 -0.0 1.57079632679
2 turn 0.0 1.0 1.0 -1.57079632679 1.57079632679 0
3 turntwist -0.0 1.0 0.0 3.14159265359 -0.0 0.0
4 turn 1.0 1.0 -0.0 -3.14159265359 -1.57079632679 0
5 turntwist 1.0 1.0 1.0 0.0 -0.0 -1.57079632679
6 turn 1.0 0.0 1.0 1.57079632679 1.57079632679 0
7 straight 1.0 0.0 -0.0 3.14159265359 -0.0 0.0
And could be loaded like this:
/dnageom/definitionFile /path/to/definition.txt # or another path
/dnageom/fractalScaling 50 50 50 nm # scale each unit to 50 nm blocks